

Examining Your BLAST Results
Review the BLAST page for each of your records by clicking the BLAST "B" on each record within your MycoMap project.


How to Interpret BLAST Results
There is not a universally applicable quantitative method to make this initial assessment for all fungal species, but a good guide involves using four components of the reference sequences in BLAST results:
- high Identity value, in the ‘identity’ column;
- high "Query Cover" value, in the ‘Query’ column;
- the number of reference sequences being matched this high in the results; and
- that the species name comes from a trusted source.
As a general rule-of-thumb for species-level determination of many groups of fungi, two specimens with a sequence similarity of 97% or above are considered to be a single species, it is not a hard and fast rule. Keep in mind, however, that this number is highly variable between different groups. For some groups, two specimens may have 99% sequence similarity, but they may be from two different, related species.
For the purposes of the initial triage, a high Identity value generally falls in the range of 98-100% sequence similarity. The identity reports on the percentage of base pairs that are the same between the sequence of your specimen and that of the reference specimen. If 99 out of 100 base pairs match, then you have a 99% identity value in your results. If there are a large number of reference sequences that fall into the 98-100% range in your results, all with the same species name you believed it your specimen to be, then you would likely identify your specimen as that species, and not need to review that sequence much more in the future. If more than one reference species have 98-100% sequence similarity with your specimen, you would identify your specimen conclusively at the genus level, but not at a species level.
Query cover is the percentage of the query sequence (your specimen) that overlaps the reference sequence. BLAST results do not typically attempt to match the full length of a sequence. A high Query Cover value for the initial triage is in the 70%+ range. If the top results fall below this range, it would generally be a good idea to review the sequence more in the future, and not verify it as a part of your initial triage. Often, the results only report on a single segment of a sequence that most closely aligns. It is possible to have a 99% identity match, but only across 35% of your sequence (query cover). In this case you would have no information on how closely the other 65% of your sequence matches up. This means reviewing the query cover percentage of a BLAST search is just as important as the identity percentage, especially when performing an initial triage.
You can see this visually from your MycoMap BLAST results. NCBI stores the data for 3 days from the time the BLAST is initiated. Click the following button:
It will take you to the NCBI BLAST results page. The red lines are a visual representation of the query cover your results are based on:
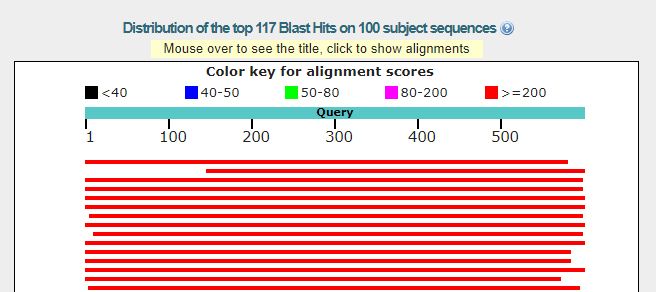
The final thing to keep in mind is that the species names associated with reference sequences in your search may not be accurate. It all comes down to how much you trust the identifier who originally proposed the name for the reference sequence. If the sequence is from a type specimen or from a paper that reviews the group, that certainly carries a lot of weight. Most reference sequences, however, will have little supporting provenance on how the species determination was made. We will discuss this more in the future, but always keep this in mind, especially when you are new to analyzing sequence data. You can view the information on the identifier of the reference sequence by clicking on the corresponding link to the reference sequence in the ‘Accession’ column.
You are inspecting the results to see if the genus and species at the top of each BLAST is the same as the name you applied to the specimen. With some of your specimens, it will be very easy to make an accurate species determination. Some records will be much more difficult. This page discusses how to approach a "first look" at the BLAST results for your specimen.
Reviewing your Results
I have a great match with just one species. What now?

Records in a project can be filtered by whether or not the display includes these assessed (verified) records.
The ultimate goal of a project owner is to verify all of the records in their projects with the "correct" species name. In you are not absolutely sure of the species name for a record, consider changing the "name-in-use" of the source record (MO or iNat) to the genus, rather than a species name you are unsure of.
Missing Sequences and/or BLAST Results
When you review your specimen records, you may find that BLAST results and sequences are not available for some specimens. Sequences may not be available if we were either:
- unable to isolate DNA from a specimen, or
- the specimen was contaminated with another fungus and we were unable to obtain a sequence.
Ensure your results do not represent a contaminant.
In a small number of cases, the sequence results may represent some kind of contaminant, such as another type of fungus like a yeast or a mold. This is an unavoidable aspect of the process, and is much more common in certain groups, such as jelly fungi. There may be a couple of reasons for this:
- The specimen may have been contaminated with another fungus in situ, i.e. where it was collected. An example may be when more than one fungus is growing on a log. Mycelium of one species may have grown into the specimen you collected.
- It is also much more common for specimens that were not immediately placed in the dryer or were otherwise dried improperly.
If you receive a sequence of a contaminant back, there is little we can do except attempt another DNA extraction from the original material. This would be an additional fee and there is no guarantee the next attempt would be successful. Typically, most researchers do not attempt this unless the specimen has some kind of unusual importance.
If you do believe the sequence to be from a contaminant, please complete the following steps:
Clickk the sequence "S" box on the line with the record you believe represents a contaminated sequence. Underneath the sequence record, you will see a gray checkbox that says "This sequence is a contaminant." Clicking the gray checkbox turns it green. Selecting this box removes the NCBI GenBank verification icon from the record, so the contaminanted sequence is not accidentally uploaded to GenBank.
We do save these sequences in the database. The may serve some type of unusual ecological information in the future.
Sequence doesn’t match the genus of your specimen
Occasionally, specimens become improperly numbered in the field or mixed up in the lab. The final sanity check for sequences is that the genus that most commonly appears in the BLAST results is the same genus that you were expecting the sequence to be. If your BLAST results show the sequence represents a Tricholoma, but the specimen is attached to an observation of Polyporus, there is obviously a problem.
The most typical issue is that two tubes were switched at some point in the process. Once you are done going through your specimens, it is usually possible to find the tube that was switched out. If you are able to find the cause of the error, the simplest fix is to edit the sequence records to be associated with the correct observational report.
If you are unable to find the cause of the error, please select the "This sequence is the wrong specimen" checkbox and alert us to the issue by emailing steve@mycomap.com. We will attempt to track down the source of the error.